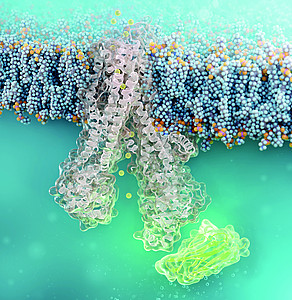
Image: Cell-penetrating nanobody (green) binds to defective CFTR chloride channel (structural simulation) © FMP | Barth van Rossum
Berlin
17 April 2026
A tiny antibody component could fundamentally change the treatment of cystic fibrosis: For the first time, researchers have succeeded in developing a so-called nanobody that can directly penetrate human cells and repair the most frequently defective chloride channel in cystic fibrosis. The new therapeutic approach was developed jointly by teams from Charité – Universitätsmedizin Berlin and the Leibniz Research Institute for Molecular Pharmacology (FMP). The results have now been published in the journal Nature Chemical Biology*.
Cystic fibrosis (CF) is a disease caused by genetic defects in the CFTR channel. This channel regulates the transport of water and salt in the lung lining and ensures the production of sufficient mucus. Approximately 90 percent of cystic fibrosis patients have a mutation in the CFTR channel known as F580del, meaning that a single amino acid is missing at position 508 in its protein chain. This alteration causes CFTR to misfold and be prematurely degraded inside the cell, instead of functioning as a channel in the cell membrane of the airways. As a result, affected individuals have thick mucus in their lungs, and pathogens can no longer be effectively cleared. This leads to chronic infection and inflammation of the airways, resulting in a progressive loss of lung function – in the worst case, necessitating a lung transplant.
Professor Marcus Mall, Director of the Department of Pediatrics with a focus on Pneumology, Immunology, and Intensive Care Medicine at Charité, and his team have made significant contributions in recent years to noticeably improving the treatment of cystic fibrosis through therapy with three small-molecule drugs (CFTR modulators): Using triple therapy with elexacaftor, tezacaftor, and ivacaftor (ETI), the function of the CFTR channel can be increased to approximately 50 percent of its normal value. However, chronic inflammation and infection of the lungs often persist, and there are also patients for whom this therapy is ineffective or who cannot tolerate it.
An antibody as a repair aid
For this group, further treatment options may become available in the future: The team led by chemist Prof. Christian Hackenberger at the Leibniz-FMP has developed a new molecule in the laboratory that stabilizes the misfolded CFTR directly inside the cell. This molecule is a nanobody – a tiny but stable antibody component that can precisely bind to defined protein surfaces. It is chemically equipped with a “transport signal,” so-called cell-penetrating peptides, which help it to enter the mucosal cells of the lungs. There, the nanobody binds to the defective channel protein and helps it assume the correct shape.
The researchers were able to demonstrate that the nanobody adhered to the mutated CFTR channel in cells derived from cystic fibrosis patients for at least 24 hours. It did not damage the cells. Functional studies also confirmed that the corrected channel once again transported chloride across the cell membrane.
Combination of Triple Therapy and Nanobody
In combination with the established ETI triple therapy, the nanobody showed a pronounced synergistic effect in these cell cultures: While the ETI agents restored the function of the defective CFTR channel by approximately half on average, the channel activity could be increased to almost 90 percent of the normal level by the additional administration of the nanobody.
This study thus demonstrates that externally administered, cell-permeable nanobodies can stabilize disease-relevant, misfolded proteins inside cells and restore their function. “Besides demonstrating the preclinical feasibility of CFTR channel repair, this is the first example of a functional, cell-permeable antibody: Until now, cell-permeable nanobodies have primarily been used to visualize intracellular target structures or to selectively kill cells,” says Christian Hackenberger.
This is not only the preclinical proof of concept for CFTR channel repair, but also the first example of a functional, cell-permeable antibody. “Because the nanobodies bind directly to the F508del mutation site, they allow for even more targeted treatment of the CFTR channel maturation disorder,” says Marcus Mall. “This new mechanism of action, in combination with existing CFTR modulators, significantly improves the correction of CFTR function. Our results suggest that this new approach could even lead to a complete normalization of CFTR function. This would be another breakthrough for cystic fibrosis therapy.” This work opens up new possibilities for further improving the treatment of cystic fibrosis—and simultaneously lays the foundation for broader therapeutic applications.
Perspectives Beyond Cystic Fibrosis
However, before this approach can be clinically applied to cystic fibrosis, key questions still need to be answered, such as developing a suitable formulation for inhalation and ensuring efficient penetration of the viscous CF mucus. Furthermore, it remains unclear how the nanobody functions in the body and how the immune system responds to nanobody treatment. These challenges are currently being addressed within the Collaborative Research Center 1449 “Dynamic Hydrogels at Biointerfaces,” under whose auspices the current results were obtained.
The approach of intracellular nanobody therapy could also be beneficial beyond cystic fibrosis for other rare genetic diseases in which protein misfolding plays a role and for which there are currently few effective treatments.
*Franz L et al. A Cell-Permeable Nanobody to Restore F508del Cystic Fibrosis Transmembrane Conductance Regulator Activity. Nat Chem Biol 2026 Apr 17. doi: 10.1038/s41589-026-02199-w
About Cystic Fibrosis
Cystic fibrosis is one of the most common fatal inherited diseases worldwide. In Germany, up to 8,000 children, adolescents, and adults are affected. Due to a disruption in the body’s salt and water balance, people with cystic fibrosis produce a thick mucus that damages organs such as the lungs and pancreas. This leads to a progressive loss of lung function and shortness of breath, which still significantly reduces life expectancy despite improved treatment approaches. Approximately 150 to 200 children are born with this rare disease in Germany each year. Testing for cystic fibrosis is part of newborn screening.
SOURCE: Joint press release from Charité and FMP
Leave a Reply