Berlin, 8 February 2023
Researchers at the Charité – Universitätsmedizin Berlin, the Max Planck Institute for Molecular Genetics (MPIMG) and the University Hospital Schleswig-Holstein (UKSH) have clarified in detail how the extremely rare hereditary BPTA syndrome develops: The change in the charge of a protein upsets the cellular self-organisation, and a developmental disorder is the result. The team also identified hundreds of comparable genetic alterations that are associated with disorders of brain development or a predisposition to cancer, among other things. The mechanism now described in the scientific journal Nature* could thus be the cause of numerous unexplained diseases.
Thousands of genetic mutations are associated with various diseases. However, it is usually unclear how exactly these mutations lead to diseases. This is because they affect sections of proteins whose three-dimensional structure is disordered and about whose function in the cell little is known so far. “The tasks of such protein sections are difficult to research because they often only unfold their effect together with other molecules,” says Dr. Martin Mensah from the Institute of Medical Genetics and Human Genetics at the Charité. He is one of the two first authors of the study and a fellow of the Digital Clinician Scientist Programme, which Charité runs together with the Berlin Institute of Health at Charité (BIH). “Using BPTA syndrome as an example, we have now described in detail how changes in disordered protein regions can cause a genetic disease.” The research team has thus discovered a new mechanism for the development of hereditary diseases – which, according to the study, is surprisingly not that rare.
BPTA stands for “brachyphalangia polydactyly and tibial aplasia/hypoplasia”. Affected individuals have severe deformities of the limbs, face, nervous and bone systems and other organs. With fewer than ten documented cases worldwide, the disease is extremely rare. To find the cause of BPTA syndrome, the researchers decoded the genetic information of five affected individuals and discovered that the protein HMGB1 is altered in all of them: the last third of its structure is no longer negatively but positively charged due to a so-called Raster shift mutation.
The nucleus solidifies
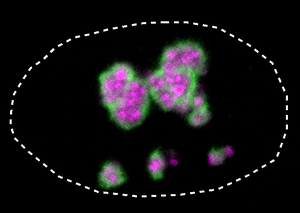
Due to the change in charge, HMGB1 now resembles proteins that prefer to reside in the so-called nuclear corpuscle. The nuclear corpuscle is a small area in the cell nucleus where parts of the protein factories are assembled. It is therefore fundamentally important for the viability of a cell. As the research team demonstrated using experiments with isolated proteins and cell cultures, the mutated HMGB1 protein is mistakenly attracted to the nuclear corpuscle with its now positively charged tail. Moreover, because the protein extension has also become partially tougher, the HMGB1 protein clumps together. “Under the microscope, we could see that the nuclear corpuscle loses its liquid-like properties and increasingly solidifies,” explains Dr Henri Niskanen, a scientist at the MPIMG and lead author of the study.
The solidification of the nuclear corpuscle impairs the vital function of the cells: More cells died in the culture with the mutated protein than without the mutation. Prof. Dr. Malte Spielmann, Director of the Institute of Human Genetics at UKSH and one of the three lead authors of the study, sums up: “We have thus shown how mutations in disordered protein segments can cause a disease: Due to a change in charge, the protein mistakenly accumulates in the nuclear corpuscle and thus impairs its vital function. As a result, the organism’s development is disturbed.”
Existing diseases explained in a new way
The research team then searched the DNA sequences of thousands of individuals in databases looking for similar cases. In fact, the scientists were able to identify more than 600 mutations in 66 proteins that gave the protein tail both a positive charge and tougher properties. 101 of these had previously been linked to various diseases, including neuronal developmental disorders and increased susceptibility to cancer. For 13 selected proteins, the team tested in cell culture whether the mutations gave them an affinity for the core corpuscle. This was true in twelve cases. About half of the tested proteins impaired the function of the nuclear corpuscle and thus resembled the disease mechanism of BPTA syndrome.
“The developmental mechanism we discovered in BPTA syndrome could therefore be used in many other diseases,” says Prof. Dr. Denise Horn, lead study author from the Institute of Medical Genetics and Human Genetics at Charité. “We are thus opening a door that could lead to the elucidation of numerous other diseases. The real work is therefore only beginning now.”
At least for some diseases, the now known mechanism could also provide a new therapeutic approach. “Tumour diseases are due to genetic changes in the affected cells,” explains Dr Denes Hnisz, research group leader at the MPIMG and third lead author of the study. “So, in the future, we may be able to stop cancer development by interfering with the cell’s self-organisation, which is mediated by disordered protein stretches.”
*Mensah MA, Niskanen H et al. Aberrant phase separation and nucleolar dysfunction underlie rare diseases. Nature 2023 Feb 08. doi: 10.1038/s41586-022-05682-1
Caption: Nucleus (dotted line) of a human cell. The mutated HMGB1 protein (green) in BPTA syndrome forms a hard layer on the nuclear corpuscle (pink) and thus causes the developmental disorder © MPIMG | Henri Niskanen
Links:
Institut für Medizinische Genetik und Humangenetik der Charité
Max-Planck-Institut für molekulare Genetik
Institut für Humangenetik des Universitätsklinikums Schleswig-Holstein